



Forma Farmacéutica y Formulación
Forma Farmacéutica
Tableta de liberación prolongada
Composición Cualitativa y Cuantitativa
astik® SR 75 mg Tabletas de liberación prolongada Cada tableta de liberación prolongada contiene:
Pregabalina…75 mg
Excipientes c.s.p.
astik® SR 150 mg Tabletas de liberación prolongada Cada tableta de liberación prolongada contiene:
Pregabalina…150 mg
Excipientes c.s.p.
astik® SR 300 mg Tabletas de liberación prolongada Cada tableta de liberación prolongada contiene:
Pregabalina…300 mg
Excipientes c.s.p.
Descripción
Nombre Comercial
astik® SR 75 mg Tabletas de liberación prolongada
astik ® SR 150 mg Tabletas de liberación prolongada
astik ® SR 300 mg Tabletas de liberación prolongadaNombre Genérico
Pregabalina
Código ATC
N03AX16
N: Sistema nervioso
N03: Antiepilépticos
N03A: Antiepilépticos
N03AX: Otros antiepilépticos
N03AX16: Pregabalina
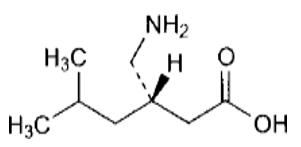
Estructura
Nombre Químico
Pregabalina
Ácido (S)-3- (aminometil)-5-metilhexanoico
PROPIEDADES FARMACOLÓGICAS
a. Propiedades farmacodinámicas
El principio activo, pregabalina, es un análogo del ácido gamma- aminobutírico [ácido (S)-3-(aminometil)-5-metilhexanoico].
Mecanismo de acción
La pregabalina se une a una subunidad auxiliar (proteína α2-δ) de los canales de calcio dependientes del voltaje en el Sistema Nervioso Central.
Eficacia clínica y seguridad
Dolor neuropático
Se ha demostrado la eficacia en ensayos clínicos en neuropatía diabética, neuralgia postherpética y lesión de la médula espinal. No se ha estudiado la eficacia en otros modelos de dolor neuropático.
La pregabalina se ha estudiado en 10 ensayos clínicos controlados con una duración de hasta semanas y dos administraciones al día (DVD) y con una duración de hasta 8 semanas y tres administraciones al día (TVD). En términos generales, los perfiles de seguridad y eficacia para los regímenes posológicos de dos y tres veces al día fueron similares.
En ensayos clínicos de hasta 12 semanas de duración para dolor neuropático periférico y central, se observó una reducción del dolor a la primera semana de tratamiento y se mantuvo a lo largo del período de tratamiento.
En ensayos clínicos controlados para dolor neuropático periférico, el 35% de los pacientes tratados con pregabalina y el 18% de los pacientes con placebo experimentaron una mejoría de un 50% en la escala de dolor. En el caso de los pacientes que no experimentaron somnolencia, dicha mejoría se observó en un 33% de los pacientes tratados con pregabalina y en un 18% de los pacientes con placebo. En el caso de los pacientes que experimentaron somnolencia, los porcentajes de respondedores fueron del 48% para pregabalina y 16% para placebo.
En el ensayo clínico controlado para dolor neuropático central, el 22% de los pacientes tratados con pregabalina y el 7% de los pacientes con placebo experimentaron una mejoría del 50% en la escala de dolor.
Epilepsia
Tratamiento complementario
La pregabalina se ha estudiado en 3 ensayos clínicos controlados con una duración de hasta 12 semanas tanto con la administración DVD como con TVD. En términos generales, los perfiles de seguridad y eficacia para los regímenes posológicos de dos y tres veces al día fueron similares. Se observó una reducción en la frecuencia de las crisis a la primera semana de tratamiento.
Población pediátrica
No se ha establecido la eficacia ni la seguridad de pregabalina como tratamiento complementario para la epilepsia en pacientes pediátricos de menos de 12 años y adolescentes. Los acontecimientos adversos observados en un estudio de farmacocinética y tolerabilidad en el que participaron pacientes de entre 3 meses y 16 años de edad (n = 65) fueron similares a los observados en los adultos. Los resultados de un estudio de seguridad, sin enmascaramiento, de 1 año de duración en 54 pacientes pediátricos de entre 3 meses y 16 años de edad con epilepsia indican que los acontecimientos adversos de pirexia e infecciones respiratorias altas se observaron con mayor frecuencia que en los estudios en adultos.
Monoterapia (pacientes recientemente diagnosticados)
Pregabalina se ha estudiado en 1 ensayo clínico controlado de 56 semanas de duración administrada DVD. Pregabalina no demostró no inferioridad frente a lamotrigina en base a la variable de estar libre de crisis durante 6 meses. Pregabalina y lamotrigina tuvieron perfiles de seguridad similares y buena tolerabilidad.
Trastorno de ansiedad generalizada
La pregabalina se ha estudiado en 6 ensayos controlados de 4-6 semanas de duración, un estudio en pacientes de edad avanzada de 8 semanas de duración y un estudio a largo plazo de prevención de recaídas con una fase doble ciego de prevención de recaídas de 6 meses de duración.
En la primera semana se observó un alivio de los síntomas del TAG como se reflejó en la Escala de Valoración de la Ansiedad de Hamilton (HAM-A).
En los ensayos clínicos controlados (4-8 semanas de duración) el 52% de los pacientes tratados con pregabalina y el 38% de los que recibieron placebo mejoraron la puntuación total de la HAM-A en al menos un 50% desde la visita basal hasta la finalización del estudio.
En ensayos clínicos controlados, una mayor proporción de pacientes tratados con pregabalina, en comparación con aquellos tratados con placebo, notificó visión borrosa que en la mayoría de los casos se resolvió al continuar con el tratamiento. Se realizaron pruebas oftalmológicas (incluyendo pruebas de agudeza visual, pruebas de campo visual y examen fundoscópico en pupila dilatada) a más de 3.600 pacientes como parte de los ensayos clínicos controlados. La agudeza visual se redujo en un 6,5% en los pacientes tratados con pregabalina frente al 4,8 % en los pacientes tratados con placebo.
Se detectaron alteraciones del campo visual en el 12,4 % de los pacientes tratados con pregabalina frente al 11,7% de los pacientes tratados con placebo. Se observaron cambios fundoscópicos en el 1,7% de los pacientes tratados con pregabalina frente al 2,1% de los pacientes tratados con placebo.
b. Propiedades farmacocinéticas.
Los parámetros farmacocinéticos de pregabalina en el estado estacionario son similares en voluntarios sanos, pacientes con epilepsia recibiendo fármacos antiepilépticos y pacientes con dolor crónico.
Absorción
La pregabalina se absorbe rápidamente cuando se administra en ayunas, alcanzando concentraciones plasmáticas máximas una hora tras la administración tanto de dosis única como de dosis múltiples. La biodisponibilidad oral de pregabalina se estima que es 90% y es independiente de la dosis. Tras la administración repetida, el estado estacionario se alcanza en las 24 a 48 horas posteriores. La velocidad de absorción de pregabalina disminuye cuando se administra con alimentos, produciéndose un descenso en la Cmax de aproximadamente un 25-30% y un retraso en el tmax de aproximadamente 2,5 horas. Sin embargo, la administración de pregabalina junto con alimentos no tiene ningún efecto clínicamente significativo sobre el grado de absorción de pregabalina.
Distribución
En estudios preclínicos, se ha visto que la pregabalina atraviesa la barrera hematoencefálica en ratones, ratas y monos. Se ha visto que la pregabalina atraviesa la placenta en ratas y está presente en la leche de ratas lactantes. En humanos, el volumen de distribución aparente de la pregabalina tras la administración oral es de aproximadamente 0,56 l/kg. La pregabalina no se une a las proteínas plasmáticas.
Biotransformación
La pregabalina sufre un metabolismo insignificante en humanos. Tras una dosis de pregabalina marcada isotópicamente, aproximadamente el 98% de la radioactividad recuperada en orina procedía de pregabalina inalterada. El derivado N-metilado de pregabalina, metabolito principal de ésta encontrado en orina, representó el 0,9% de la dosis. En estudios preclínicos, no hubo indicios de racemización del S-enantiómero de pregabalina al R- enantiómero.
Eliminación
La pregabalina se elimina del sistema circulatorio principalmente mediante excreción renal como fármaco inalterado. La semivida media de eliminación de pregabalina es de 6,3 horas.
El aclaramiento plasmático y el aclaramiento renal de pregabalina son directamente proporcionales al aclaramiento de creatinina.
Es necesario un ajuste de la dosis en pacientes con la función renal alterada o en hemodiálisis.
Linealidad/No linealidad
La farmacocinética de pregabalina es lineal en el rango de dosis diaria recomendada. La variabilidad farmacocinética interindividual de pregabalina es baja (<20%). La farmacocinética de dosis múltiples es predecible a partir de los datos obtenidos con dosis única. Por tanto, no es necesario llevar una monitorización rutinaria de las concentraciones plasmáticas de pregabalina.
Sexo
Los ensayos clínicos indican que el sexo no tiene influencia clínicamente significativa sobre las concentraciones plasmáticas de pregabalina.
Alteración renal
El aclaramiento de pregabalina es directamente proporcional al aclaramiento de creatinina. Además, la pregabalina se elimina del plasma de forma eficaz mediante hemodiálisis (tras una sesión de hemodiálisis de 4 horas, las concentraciones plasmáticas de pregabalina se reducen aproximadamente al 50%). Dado que la eliminación por vía renal es la principal vía de eliminación, en pacientes con insuficiencia renal es necesaria una reducción de la dosis y una dosis complementaria tras la hemodiálisis.
Alteración hepática
No se han llevado a cabo estudios de farmacocinética específicos en pacientes con la función hepática alterada. Puesto que la pregabalina no sufre un metabolismo significativo y se excreta mayoritariamente como fármaco inalterado en orina, no es previsible que una alteración de la función hepática altere de forma significativa las concentraciones plasmáticas de pregabalina.
Población pediátrica
En un estudio de farmacocinética y tolerabilidad se evaluó la farmacocinética de pregabalina en pacientes pediátricos con epilepsia (grupos de edad: de 1 a 23 meses, de 2 a 6 años, de 7 a 11 años y de 12 a 16 años) con
concentraciones de dosis de 2,5, 5, 10 y 15 mg/kg/día.
En general, el tiempo transcurrido hasta alcanzar la concentración plasmática máxima tras la administración oral de pregabalina a pacientes pediátricos en ayunas fue similar en todo el grupo de edad y se produjo entre 0,5 horas y 2 horas después de la dosis.
Los parámetros de Cmax y AUC de pregabalina aumentaron de forma lineal con el aumento de la dosis en cada grupo de edad. El AUC fue un 30% menor en los pacientes pediátricos con un peso inferior a 30 kg debido a un mayor aclaramiento ajustado al peso corporal del 43% en estos pacientes en comparación con los pacientes con un peso ≥30 kg.
La semivida terminal promedio de pregabalina fue, aproximadamente, de entre 3 y 4 horas en los pacientes pediátricos de hasta 6 años de edad, y de entre 4 y 6 horas en los de 7 años o más.
El análisis de farmacocinética poblacional mostró que el aclaramiento de creatinina era una covariable significativa del aclaramiento de pregabalina oral, el peso corporal era una covariable significativa del volumen de distribución aparente de pregabalina oral, y que dichas relaciones eran similares en los pacientes pediátricos y adultos.
No se ha estudiado la farmacocinética de pregabalina en pacientes de menos de 3 meses de edad.
Pacientes de edad avanzada
El aclaramiento de pregabalina tiende a disminuir al aumentar la edad. Este descenso en el aclaramiento de pregabalina oral está en relación con el descenso del aclaramiento de creatinina asociado con el aumento de la edad. Podría requerirse una reducción de la dosis de pregabalina en pacientes que tengan la función renal alterada debido a la edad.
Madres lactantes
Se evaluó la farmacocinética de 150 mg de pregabalina administrados cada 12 horas (dosis diaria de 300 mg), en 10 mujeres lactantes, tras al menos 12 semanas después del parto. La lactancia tuvo un efecto nulo o pequeño sobre la farmacocinética de pregabalina. Pregabalina se excretó por la leche materna a concentraciones promedio, en estado de equilibrio, de aproximadamente el 76% de las presentes en el plasma materno. La dosis estimada para el lactante procedente de la leche materna (suponiendo un consumo medio de leche de 150 ml/kg/día) de las mujeres que reciben 300 mg/día o la dosis máxima de 600 mg/día sería de 0,31 o 0,62 mg/kg/día, respectivamente. Estas dosis estimadas son aproximadamente el 7% de la dosis materna diaria total, en mg/kg.
DATOS CLÍNICOS
a. Indicaciones terapéuticas
Tratamiento para el dolor neuropático.
Dolor neuropático asociado con neuropatía periférica diabética. Epilepsia (crisis parciales con o sin generalización secundaria). Fribromialgia.
Desórdenes de ansiedad. Neuralgia postherpética.
b. Posología y forma de administración Vía de Administración: Vía oral.
astik® SR 75/150/300 mg es administrado por vía oral con o sin alimentos.
La dosis recomendada es una tableta diaria de astik® SR 75/150 mg diarios y después de una semana (dependiendo de la respuesta) es posible aumentar la dosis según sea necesario a una tableta de astik® SR 300 mg con o sin alimentos. La dosis máxima de astik® SR es de 600 mg /día.
Dolor neuropático
La dosis debe de comenzar con una tableta de 150 mg/día y puede incrementar después de 3 – 7 días (de acuerdo a la respuesta) a una tableta de 300 mg / día. Finalmente, si la respuesta observada no es la deseada, puede incrementarse después de 7 días a 600 mg/día basados en la tolerancia y la eficacia.
Dolor neuropático asociado con neuropatía periférica diabética:
La dosis debe comenzar con una tableta de 150 mg/día y puede incrementarse después de una semana a una dosis máxima recomendada de astik® SR 300 mg/día en pacientes con aclaramiento de creatinina de al menos 60 mL/min.
Aunque la pregabalina también fue estudiada a 600 mg/día, no hay evidencia que esta dosis confiera beneficios adicionales significativos y esta dosis fue menos tolerada. En vista de las reacciones adversas dependientes de la dosis, el tratamiento con dosis arriba de 300 mg/día no está recomendado.
Neuralgia postherpética: La dosis recomendada de astik® SR 75/150/300 mg es 150 mg a 300 mg por día en pacientes con aclaramiento de creatinina de 30 – 60 mL/min. La dosis debe de comenzar con una tableta de 150 mg por día y puede incrementarse a una tableta de 300 mg/día en una semana basado en la eficacia y la tolerabilidad. Pacientes que no experimenten suficiente alivio del dolor en las siguientes 2 ó 4 semanas de tratamiento y tengan un aclaramiento de creatinina mayor de 60 ml/min pueden incrementar la dosis a 600 mg/día.
En vista de las reacciones adversas dependientes de la dosis y un alto índice de discontinuidad del tratamiento debido a las reacciones adversas, dosis arriba de 300 mg/día deben ser reservadas únicamente para aquellos pacientes que siguen teniendo dolor y están tolerando los 300 mg diariamente.
Epilepsia (crisis parciales con o sin generalización secundaria). Iniciar con 150 mg/día. Después de 1 semana puede incrementarse la dosis a 300 mg/día. Dependiendo de la respuesta puede incrementarse a una dosis máxima de 600 mg/día, después de 2 semanas.
Trastorno de ansiedad generalizada. Iniciar con 150 mg/día. Después de 1 semana de tratamiento puede incrementarse a intervalos semanales de 300 mg/día. Dosis máxima 600 mg/día.
Fibromialgia: La dosis inicial es de 150 mg diarios. Después de 1 semana de tratamiento y de acuerdo a la respuesta observada, puede incrementarse la dosis a 300 mg diarios y luego puede incrementarse la dosis a 450 mg/día si es necesario.
Pacientes con insuficiencia renal: astik® SR 75/150/300 mg no es adecuado para pacientes con insuficiencia renal.
Pacientes con insuficiencia hepática: debido a que pregabalina no sufre un metabolismo hepático significativo y se excreta predominantemente como fármaco inalterado en la orina, no se requiere ajuste de dosis en pacientes con insuficiencia hepática.
Recomendación a Población Pediátrica: No se ha establecido seguridad y eficacia en prebabalina en niños menores de 12 años ni en adolescentes (de 12 a 17 años de edad).
Pacientes de edad avanzada: Los pacientes de edad avanzada pueden precisar una reducción de la dosis de pregabalina debido a la disminución de la función renal.
Tabla de dosis en caso de pacientes con insuficiencia renal de acuerdo con el grado de aclaramiento de creatinina
La pregabalina se elimina del sistema circulatorio principalmente por excreción renal como fármaco inalterado. Dado que el aclaramiento plasmático de pregabalina es direrectamente proporcional al aclaramiento de creatinina, la reducción de la dosis en pacientes con la función renal alterada se deberá realizar de forma individualizada de acuerdo con el aclaramiento de creatinina (Acr), tal y como se indica en la tabla 1, que se ha determinado usando la fórmula siguiente:
Acr (ml/min) [1.23x (140- edad (años) x peso (Kg) / creatinina sérica (µ mol/ L] (x 0.85 si se trata de una mujer)
La pregabalina se elimina del plasma de forma eficaz mediante hemodiálisis (50% del fármaco en 4 horas). En pacientes sometidos a hemodiálisis, se debe ajustar la dosis diaria de pregabalina según su función renal. Además de la dosis diaria, después de cada sesión de 4 horas de hemodiálisis se debe administrar de forma inmediata una dosis complementaria.
Tabla de ajuste de dosis de acuerdo a la función renal
| Aclaramiento de creatinina (Acr) (ml/min) | Dosis diaria total de pregabalina * | Posología | |
| Dosis inicial (mg/día) | Dosis máxima (mg/día) | ||
| ≥ 60 | 150 | 600 | DVD o TVD |
| ≥ 30 -<60 | 75 | 300 | DVD o TVD |
| ≥ 15 – < 30 | 25-50 | 75 | UVD |
| < 15 | 25 | ||
| Dosis complementaria tras la hemodiálisis (mg) | |||
| 25 | 100 | Dosis única | |
TVD = Tres veces al día DVD= Dos veces al día UVD = Una vez al día
*La dosis diaria total (mg/día) se debe dividir en la toma indicada en la posología para obtener los mg/dosis adecuados.
+ La dosis complementaria es una única dosis adicional.
c. Contraindicaciones
astik® SR está contraindicado en pacientes con hipersensibilidad conocida a la pregabalina o cualquiera de sus otros componentes.
d. Advertencias y precauciones especiales de empleo
Angioedema: Han habido reportes postcomercialización de angioedema en pacientes durante el tratamiento inicial y crónico con pregabalina. Síntomas específicos incluyeron hinchazón de la cara, boca (lengua, labios y encías), y cuello (garganta y laringe). Hubo reportes de angioedema con amenaza de la vida con compromiso respiratorio que requirieron tratamiento de emergencia. Pregabalina debe ser descontinuada inmediatamente en pacientes con esos síntomas. Precauciones deben ser aplicadas cuando se prescribe pregabalina a pacientes que han tenido episodios previos de angioedema. Además, pacientes que están tomando otros medicamentos asociados con angioedema (ej.: inhibidores de la enzima convertidora de angiotensina [Inhibidores de ACE]) pueden tener un aumento de riesgo de desarrollar angioedema.
Hipersensibilidad: Han habido reportes postcomercialización de hipersensibilidad en pacientes poco después de iniciar el tratamiento con pregabalina. Reacciones adversas incluyeron enrojecimiento de la piel, ampollas, urticaria, erupciones, disnea y sibilancias. Pregabalina debe ser descontinuada inmediatamente en pacientes que presentan estos síntomas.
Ideación y comportamiento suicida: Pregabalina aumenta el riesgo de pensamientos o comportamientos suicidas en pacientes que toman estos medicamentos. Pacientes en tratamiento con pregabalina deben ser monitoreados por la aparición o empeoramiento de la depresión, pensamientos o comportamientos suicidas, y/o cualquier cambio inusual en el estado de ánimo o comportamiento. Cualquiera que considere prescribir pregabalina debe balancear el riesgo de pensamientos o comportamientos suicidas con el riesgo de una enfermedad no tratada. Si aparecen pensamientos y comportamientos suicidas durante el tratamiento, el médico necesita considerar si la aparición de estos síntomas en cualquier paciente pueden estar relacionados a la enfermedad siendo tratada. Los pacientes, sus cuidadores y las familias deben ser informados de que pregabalina aumenta el riesgo de pensamientos y comportamientos suicidas, y deben ser aconsejados de la necesidad de estar alertas a la aparición o empeoramiento de las señales y síntomas de depresión, cualquier cambio inusual en el estado de ánimo o comportamiento, o en el aparecimiento de pensamientos suicidas, comportamientos, o pensamientos acerca de autolesiones.
Comportamientos preocupantes deben de ser reportados inmediatamente a los profesionales de la salud.
Edema periférico: El tratamiento con pregabalina puede causar edema periférico. En estudios de corto plazo pruebas en pacientes sin enfermedades de corazón o vascular periféricas clínicamente relevantes, no hubo asociación aparente entre el edema periférico y complicaciones cardiovasculares como hipertensión o insuficiencia cardíaca congestiva. El edema periférico no fue asociado a cambios de laboratorio indicativos de deterioro de la función renal o hepática. Una más alta frecuencia de aumento en peso y edema periférico fue observada en pacientes tomando juntos pregabalina y agentes antidiabéticos tiazolidinedionas comparado con pacientes tomando solamente una de estas dos. Asi como la tiazolidinedionas un tipo de medicamentos antidiabéticos puede causar aumento de peso y/o retención de fluidos, posiblemente exacerbando o conduciendo a la insuficiencia cardíaca, se debe tener cuidado cuando se coadministra pregabalina y estos agentes.
Aumento en peso: El tratamiento con pregabalina puede causar aumento de peso. La asociación al aumento de peso y pregabalina fue relacionada a la dosis y duración de exposición, pero no parece estar asociado con la línea basal de IMC, género o edad. El aumento de peso no se limitó a pacientes con edema. Aunque el aumento en peso no se asoció con cambios clínicamente importantes en la presión arterial en estudios controlados a corto plazo, los efectos cardiovasculares a largo plazo de pregabalina asociados al aumento de peso son desconocidos, mientras que los efectos de la pregabalina asociados al aumento de peso en el control glucémico no se han evaluado sistemáticamente. El tratamiento con pregabalina no parece estar asociado con pérdida del control glucémico (como el medido por la HbA1C).
Discontinuación abrupta o rápida: Seguido de una discontinuación rápida o abrupta de pregabalina, algunos pacientes reportaron síntomas incluyendo insomnio, náuseas, dolor de cabeza, y diarrea. Pregabalina debe disminuirse gradualmente con un mínimo de 1 semana y no interrumpido abruptamente.
Potencial tumorigénico: En estudios preclínicos estándar de carcinogenicidad in vivo del tiempo de vida, un inesperado alto índice de hemangiosarcoma fue identificado en dos diferentes variedades de ratones. El significado clínico de este descubrimiento es desconocido.
Efectos oftalmológicos: En estudios controlados, una proporción más alta de pacientes tratados con pregabalina reportaron visión borrosa que los pacientes tratados con placebo, que se resolvió en la mayoría de los casos con la continuación de la dosis. Menos de 1% de los pacientes descontinuaron el tratamiento con pregabalina debido a eventos relacionados con la visión (principalmente visión borrosa).
Aunque la importancia clínica de los hallazgos oftalmológicos son desconocidos, los pacientes deben ser informados que si ocurren cambios en la visión, deben de notificarlo a su médico. Si la alteración de la visión persiste, se debe considerar evaluación adicional. Deberá considerarse una evaluación más frecuente a los pacientes que ya está siendo rutinariamente monitoreados por condiciones oculares.
Elevación de creatina cinasa: El tratamiento con pregabalina fue asociado al aumento de la creatina cinasa. Tres pacientes tratados con pregabalina tuvieron reacciones notificadas como rabdomiólisis en los ensayos clínicos previos a la comercialización. Los pacientes deben ser instruidos a reportar con prontitud inexplicables dolores musculares, sensibilidad o debilidad, particularmente si esos síntomas musculares están acompañados de malestar o fiebre. El tratamiento con pregabalina se debe descontinuar si se diagnostica o sospecha miopatía o si ocurre un aumento marcado en los niveles de creatina cinasa.
Disminución del recuento de plaquetas: El tratamiento con pregabalina fue asociado con disminución en el recuento de plaquetas. En ensayos controlados aleatorios, la pregabalina no se asoció con un aumento de las reacciones adversas relacionadas con sangrado.
Prolongación del intervalo PR: El tratamiento con pregabalina fue asociado con la prolongación del intervalo PR. Esto no fue asociado con un aumento en el riesgo de reacciones adversas de bloqueo AV de segundo o tercer grado. Los análisis de subgrupos no identificaron un incremento del riesgo de prolongación de PR en pacientes con línea basal de prolongación PR o en pacientes tomando otros medicamentos que prolongan el PR. Sin embargo estos análisis no pueden ser considerados definitivos por el número limitado de pacientes en esta categoría.
Dermatopatía: Los pacientes diabéticos deben ser instruidos para prestar atención particularmente a la integridad de la piel mientras son tratados con pregabalina.
Fertilidad masculina: Los hombres bajo tratamiento con pregabalina que planean tener un hijo deben ser informados del riesgo potencial de teratogenicidad mediada masculina.
Falla cardiaca congestiva: Durante la experiencia postcomercialización se han notificado casos de insuficiencia cardiaca congestiva en algunos pacientes con pregabalina. Estas reacciones se observan sobre todo en pacientes de edad avanzada (mayores de 65 años) con función cardiovascular comprometida y tratados con pregabalina en la indicación de tratamiento del dolor neuropático. Pregabalina debe utilizarse con precaución en este tipo de pacientes. Estas reacciones pueden revertir tras la suspensión del tratamiento.
Falla renal: Se han notificado casos de insuficiencia renal, de los cuales algunos se revirtieron con la interrupción del tratamiento con pregabalina.
Disminución de la funcionalidad del tracto gastrointestinal inferior: Durante el período postcomercialización se han notificado casos relacionados con la disminución de la funcionalidad del tracto gastrointestinal inferior (por ejemplo: obstrucción intestinal, íleo paralítico, estreñimiento) al administrarse pregabalina conjuntamente con medicamentos con potencial para producir estreñimiento, como los analgésicos opioides. En caso de que se vayan a administrar en combinación pregabalina y opioides, debe considerarse la utilización de medidas para evitar el estreñimiento (especialmente en mujeres y pacientes de edad avanzada).
Encefalopatía: Se han notificado casos de encefalopatía, mayoritariamente en pacientes con enfermedades subyacentes que podrían haber provocado encefalopatía.
Uso en pediatría: La seguridad y eficacia de pregabalina en pacientes pediátricos no ha sido establecida. No utilizarse en menores de 18 años.
Uso en geriatría: No se han observado diferencias substanciales en la seguridad y eficacia en pacientes de edad avanzada, con relación a los adultos más jóvenes, pero el aumento en la sensibilidad no se puede descartar. Es conocido que la pregabalina se excreta principalmente por el riñón, y el riesgo de reacción tóxica a la pregabalina puede ser mayor en pacientes con deterioro de la función renal.
Debido a que la pregabalina es eliminada principalmente por excreción renal, la dosis debe ser ajustada para pacientes de avanzada edad con insuficiencia renal.
Uso incorrecto, potencial de abuso o dependencia: Se han notificado casos de uso incorrecto, abuso o dependencia. Se debe tener precaución en pacientes con antecedentes de abuso de sustancias, y los pacientes han de ser monitorizados para detectar síntomas de uso incorrecto, abuso o dependencia con pregabalina (se han notificado casos de tolerancia, aumento de la dosis, búsqueda compulsiva del medicamento).
Información importante sobre excipientes: Este medicamento contiene manitol, puede producir un efecto laxante leve.
e. Interacciones con otros medicamentos y otras formas de interacción.
Dado que la pregabalina se excreta principalmente inalterada en orina, experimenta un metabolismo insignificante en humanos (<2% de la dosis se recupera en la orina en forma de metabolitos) y no se une a las proteínas plasmáticas, es poco probable que su farmacocinética sea afectada por otros agentes a través de interacciones metabólicas o desplazamiento en la unión a proteínas. Estudios in vitro e in vivo mostraron que la pregabalina es poco probable que participe en interacciones medicamentosas farmacocinéticas significativas.
Gabapentina: La farmacocinética de gabapentina seguida de la administración única y de dosis múltiples no fue alterada por la coadministración de pregabalina.
El grado de absorción de pregabalina no sufrió alteraciones por coadmintración de gabapentina, aunque hubo una pequeña reducción en la tasa de absorción.
Anticonceptivo oral: La coadministración de pregabalina (200 mg 3 veces por día) no tuvo efecto en el estado de equilibrio farmacocinético de noretindrona y etinilestradiol (1 mg/35 g, respectivamente) en sujetos sanos.
Oxicodona y lorazepam: Dosis orales múltiples de pregabalina fueron coadministradas con oxicodona y lorazepam. Aunque no fueron observadas interacciones farmacocinéticas, efectos aditivos sobre funcionamiento cognitivo y motor grueso se observaron cuando la pregabalina se coadministra con oxicodona. No se observaron efectos clínicos importantes en la respiración.
Fenitoína, carbamazepina, ácido valproico, y lamotrigina: El estado de equilibrio de la concentración plasmática de fenitoína, carbamazepina, y carbamazepina 10,11 epóxido, ácido valproico, y lamotrigina, no se vieron afectadas por la administración concomitante de pregabalina.
Gliburida, insulina, metformina, furosemida, tiagabina: Fármaco concomitante no tiene ningún efecto sobre la farmacocinética de pregabalina y la pregabalina no tiene ningún efecto sobre la farmacocinética del fármaco concomitante.
Depresivos del sistema nervioso central: Pacientes que requieran tratamiento concomitante con depresivos del sistema nervioso central como opiodes o benzodiacepinas deben ser informados que ellos pueden experimentar efectos adversos aditivos del SNC, como somnolencia.
Alcohol: Debe informarse a los pacientes que deben evitar el consumo de alcohol mientras están tomando pregabalina, porque pregabalina puede potenciar el deterioro de las habilidades motoras y efectos sedantes del alcohol.
f. Embarazo y lactancia.
Embarazo: No existen datos suficientes sobre la utilización de pregabalina en mujeres embarazadas.
Los estudios en animales han mostrado toxicidad reproductiva. Se desconoce el riesgo en seres humanos.
astik® SR no debería utilizarse durante el embarazo excepto si fuese claramente necesario (si el beneficio para la madre es claramente superior al riesgo potencial para el feto).
Lactancia: Pregabalina se excreta en la leche materna. No se conoce el efecto de pregabalina en recién nacidos/lactantes. Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
g. Efectos sobre la capacidad para conducir y manejar maquinaria (cuando aplique).
Mareos y somnolencia: Pregabalina puede causar mareos y somnolencia. Los pacientes deben ser informados que los mareos y somnolencia por la pregabalina pueden afectar su capacidad para llevar a cabo ciertas tareas como manejar u operar maquinaria.
Se ha notificado, durante el período postcomercialización, casos de pérdida de conocimiento, confusión y deterioro mental. Por tanto, se debe aconsejar a los pacientes que tengan precaución hasta que se familiaricen con los potenciales efectos del medicamento.
h. Reacciones adversas.
Efectos indeseables con pregabalina incluyen los siguientes:
Cuerpo como un todo: Astenia, lesiones accidentales, dolor de espalda, dolor de pecho, edema facial, infecciones, dolor de cabeza, dolor y síndrome gripal.
Desórdenes digestivos: Boca seca, estreñimiento, flatulencia, vómitos, distensión abdominal.
Trastornos metabólicos y nutricionales: Edema periférico, aumento de peso, edema, retención de líquidos.
Sistema nervioso: Mareos, somnolencia, neuropatía, ataxia, vértigo, confusión, euforia, falta de coordinación, pensamiento anormal (principalmente dificultad con la concentración/atención), temblor, marcha anormal, amnesia, nerviosismo, trastornos del habla, trastorno del equilibrio, letargo.
Trastornos musculoesqueléticos: Espasmos musculares, dolor de espalda.
Trastornos psiquiátricos: Euforia, confusión, ansiedad, desorientación, depresión.
Sistema respiratorio: Disnea, bronquitis, sinusitis, dolor faringolaríngeo.
Sentidos especiales: Visión borrosa, visión anormal.
Otras reacciones adversas observadas son las siguientes: (reacciones adversas frecuentes son aquellas que ocurren por lo menos en 1/100 pacientes; reacciones adversas no frecuentes son aquellas que ocurren en 1/100 a 1/1000 pacientes; reacciones adversas raras en menos de 1/1000 pacientes):
Cuerpo como un todo: Frecuentes: dolor abdominal, reacciones alérgicas, fibre. Poco frecuentes: Absceso, celulitis, escalofríos, malestar general, rigidez de cuello, sobredosis, dolor pélvico, reacción de fotosensibilidad. Raro: reacción anafilactoide, ascitis, granuloma, efecto de resaca, lesiones intencionales, fibrosis retroperitoneal, choque.
Desórdenes cardiovasculares: Poco frecuentes: tromboflebitis profunda, insuficiencia cardiaca, hipotensión, hipotensión postural, trastorno vascular retinal, síncope. Raras: depresión ST, fibrilación ventricular.
Desórdenes del sistema digestivo: Frecuentes: gastroenteritis, aumento del apetito. Poco frecuente: colecistitis, colelitiasis, colitis, disfagia, esofagitis, gastritis, hemorragia gastrointestinal, melena, ulceración de la boca, pancreatitis, hemorragia rectal, edema de la lengua. Raro: estomatitis aftosa, úlcera esofágica, absceso periodontal.
Trastornos del sistema hémico y linfático: Frecuentes: equimosis. Poco frecuentes: anemia, eosinofilia, anemia hipocrómica, leucocitosis, leucopenia, linfadenopatía, trombocitopenia. Raro: mielofibrosis, policitemia, disminución de protrombina, púrpura, trombocitopenia.
Trastornos metabólicos y nutricionales: Raro: disminución de la tolerancia a la glucosa, cristaluria de urato.
Frecuente: Aumento del apetito.
Poco frecuente: Anorexia, hipoglucemia.
Trastornos musculoesqueléticos: Frecuentes: artralgia, calambres en las piernas, mialgia, miastenia. Poco frecuentes: artrosis. Raras: condrodistrofia, espasmo generalizado.
Trastornos nerviosos: Frecuentes: Ansiedad, despersonalización, hipertonía, hipoestesia, disminución de libido, nistagmo, parestesia, estupor, temblores. Poco frecuentes: sueños anormales, agitación, apatía, afasia, parestesia circumoral, disartria, alucinaciones, hostilidad, hiperalgesia, hiperestesia, hipercinesia, hipocinesia, hipotonía, incremento de libido, mioclonus, neuralgia. Raro: adicción, síndrome cerebeloso, rigidez de rueda dentada, coma, delirio, alucinaciones, disautonomía, discinesia, distonía, encefalopatía, síndrome extrapiramidal, síndrome de Guillain-Barré, hipoalgesia, hipertensión intracraneal, reacción maníaca, reacción paranoide, neuritis periférica, trastornos de la personalidad, depresión psicótica, reacción esquizofrénica, trastorno del sueño, tortícolis, trismo.
Desórdenes respiratorios: Raro: apnea, atelectasia, bronquiolitis, hipo, laringismo, edema pulmonar, fibrosis pulmonar, bostezo, sensación de opresión en la garganta.
Poco frecuentes: Disnea, epistaxis, tos, congestión nasal, rinitis, ronquidos, sequedad nasal.
Trastornos de la piel y apéndices: Frecuente: prurito. Poco frecuentes: alopecia, piel seca, eccema, hirsutismo, úlceras en la piel, urticaria, rash vesículobuloso. Raro: angioedema, dermatitis exfoliativa, dermatitis liquenoide, melanosis, alteraciones de las uñas, erupción petequial, erupción purpúrica, erupción pustulosa, atrofia de piel, necrosis de piel, nódulos cutáneos, síndrome de Stevens-Johnson, nódulo subcutáneo.
Sentidos especiales: Frecuentes: conjuntivitis, diplopía, otitis media, tinnitus. Poco frecuentes: anormalidad de acomodación, blefaritis, sequedad en los ojos, hemorragia ocular, hiperacusia, fotofobia, edema de retina, pérdida del gusto, alteración del gusto. Raro: anisocoria, ceguera, úlcera corneal, exoftalmos, parálisis extraocular, iritis, queratitis, queratoconjuntivitis, miosis, midriasis, ceguera nocturna, oftalmoplejía, atrofia óptica, papiledema, parosmia, ptosis, uveítis.
Trastornos urogenitales: Frecuentes: anorgasmia, impotencia, frecuencia urinaria, incontinencia urinaria. Poco frecuentes: eyaculación anormal, albuminuria, amenorrea, dismenorrea, disuria, hematuria, cálculo renal, leucorrea, menorragia, metrorragia, nefritis, oliguria, retención urinaria, orina anormal. Raro: insuficiencia renal aguda, balanitis, neoplasia vesical, cervicitis, dispareunia, epididimitis, lactación femenina, glomerulitis, trastorno ovárico, pielonefritis.
i. Sobredosificación.
No existe un antídoto específico para pregabalina. El tratamiento es generalmente sintomático y de soporte.
Para disminuir la absorción: vaciar el estómago por emesis o lavado gástrico; precauciones usuales deben ser observadas para mantener la vía aérea.
Para reforzar la eliminación: Aunque hemodiálisis no ha sido llevada a cabo en los pocos casos de sobredosis, esto puede ser indicado por el estado clínico del paciente o en pacientes con insuficiencia renal significativa. El procedimiento estándar de hemodiálisis resulta en una depuración significativa de pregabalina (aproximadamente 50% en 4 horas).
Monitoreo: monitoreo de signos vitales y observación del estado clínico del paciente.
Abuso y Adicción. Uso incorrecto, potencial de abuso o dependencia: Se han notificado casos de uso incorrecto, abuso o dependencia. Se debe tener precaución en pacientes con antecedentes de abuso de sustancias, y los pacientes han de ser monitorizados para detectar síntomas de uso incorrecto, abuso o dependencia con pregabalina (se han notificado casos de tolerancia, aumento de la dosis, búsqueda compulsiva del medicamento).
DATOS FARMACÉUTICOS
a. Lista de Excipientes
astik® SR 75 mg Tableta de Liberación Prolongada. astik® SR 150 mg Tableta de Liberación Prolongada. astik® SR 300 mg Tableta de Liberación Prolongada.
- Hipromelosa
- Povidona
- Macrogol
- Manitol
- Bicarbonato de Sodio
- Estearato de Magnesio
- Agua purificada *eliminada en el proceso de fabricación*.
b. Incompatibilidades
No procede.
c.Período de Validez
24 meses.
d. Precauciones especiales de conservación
No mayor de 30°C.
e. Naturaleza y contenido del envase.
astik® SR 75 mg Tabletas de liberación prolongada
Farmacia:
Caja con 10 tabletas de liberación prolongada.
Caja con 30 tabletas de liberación prolongada
Muestra Médica:
Caja con 2 tabletas de liberación prolongada.
Caja con 10 tabletas de liberación prolongada.
astik® SR 150 mg Tabletas de liberación prolongada
Farmacia:
Caja con 10 tabletas de liberación prolongada.
Caja con 30 tabletas de liberación prolongada
Muestra Médica:
Caja con 2 tabletas de liberación prolongada.
Caja con 10 tabletas de liberación prolongada.
astik® SR 300 mg Tabletas de liberación prolongada
Farmacia:
Caja con 10 tabletas de liberación prolongada.
Caja con 30 tabletas de liberación prolongada
Muestra Médica:
Caja con 2 tabletas de liberación prolongada.
Caja con 10 tabletas de liberación prolongada.
No todas las presentaciones están disponibles en todos los países.
FECHA DE REVISIÓN
Febrero – 2019.
REFERENCIAS BIBLIOGRAFICAS
Agencia Española de Medicamentos. AEMPS.